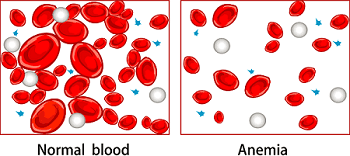
Anemia
Anemia of chronic disease Serum iron is often low in patients with chronic conditions. Reduced serum iron, which deprives bacteria of an essential nutrient necessary for growth and replication, is an immune system strategy for…
Anemia of chronic disease Serum iron is often low in patients with chronic conditions. Reduced serum iron, which deprives bacteria of an essential nutrient necessary for growth and replication, is an immune system strategy for…
